Hội chứng Alagille là gì?
Gan được cấu tạo bởi bao gan, mô gan, mạch máu và đường mật trong gan. Tế bào gan, mạch máu và đường mật trong gan tạo nên mô gan. Gan có nhiệm vụ tiết ra mật để tham gia vào quá trình tiêu hóa.
Hội chứng Alagille (ALGS) còn gọi là hội chứng thiểu sản ống mật và loạn sản động mạch gan, được mô tả lần đầu tiên vào cuối những năm 1960. Đây là một rối loạn di truyền, ngoài ảnh hưởng đến gan còn gây ra bất thường cho tim và các bộ phận khác của cơ thể. Theo đó, người mắc hội chứng Alagille có quá ít ống mật hoặc hẹp ống mật, dị dạng ống mật (ống nhỏ nối gan và ruột non) khiến mật chảy ra khỏi gan ít hơn, dẫn đến mật tích tụ trong gan và gây tổn thương gan.
Tỷ lệ mắc bệnh ước tính khoảng 1/70.000 trẻ sơ sinh và xảy ra ở cả hai giới. Một số nghiên cứu cho thấy tỷ lệ khoảng 1/30.000-1/45.000 người trong dân số nói chung.

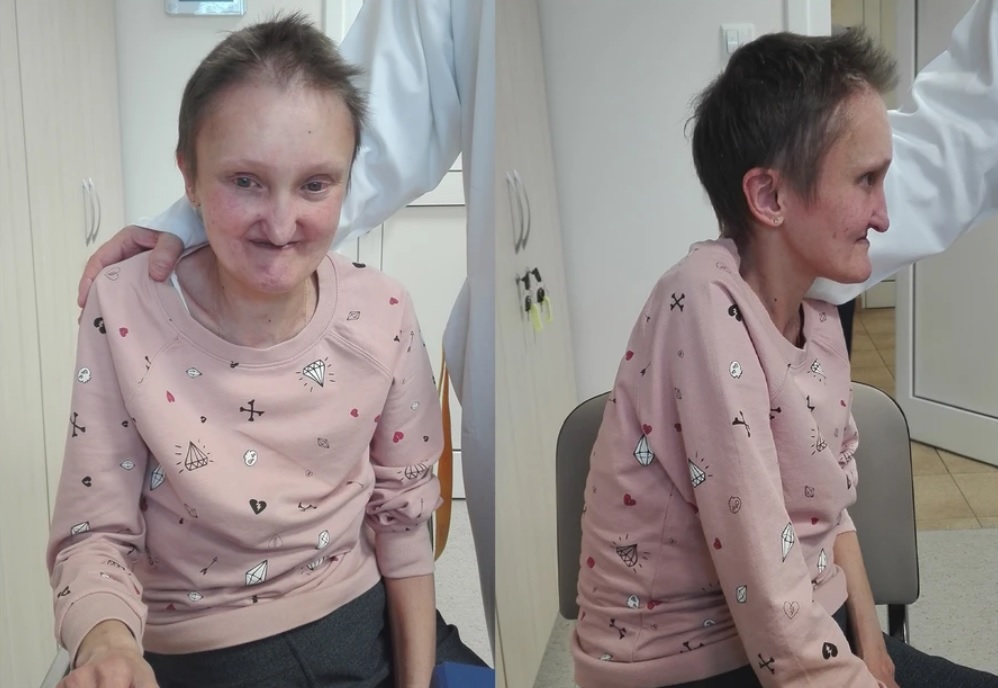
Trán cao, đôi mắt sâu, cằm nhọn và nhỏ của người mắc hội chứng Alagille (Nguồn: Di truyền y học BMC)
Nguyên nhân gây ra hội chứng Alagille
Đột biến gen JAG1 theo kiểu trội trên nhiễm sắc thể thường gây ra khoảng 90% trường hợp mắc hội chứng Alagille. Gen JAG1 nằm trên nhiễm sắc thể 20, mang thông tin tổng hợp protein Jagged-1 có liên quan đến đường truyền tín hiệu giữa các tế bào.
Một số ít trường hợp mắc hội chứng này được ghi nhận do đột biến gen NOTCH2. Gen NOTCH2 nằm trên nhiễm sắc thể số 1, cung cấp hướng dẫn để tạo ra một protein có tên là Notch2. Khi các protein Jagged-1 và Notch liên kết với nhau sẽ tạo ra các phản ứng truyền tín hiệu chi phối chức năng của tế bào, để xây dựng cấu trúc cơ thể trong phôi đang phát triển. Sai sót trong quá trình xảy ra do đột biến ở các gen này khiến cho ống mật, gan, tim bị ảnh hưởng và dẫn đến bất thường.
Các đột biến gen JAG1, NOTCH2 của trẻ mắc hội chứng Alagille có thể do cha mẹ di truyền với tỷ lệ khoảng 50%. Khoảng 50% trường hợp đột biến ở trẻ không liên quan đến DNA của cha mẹ mà tự phát trên đột biến của gen trẻ, còn gọi là đột biến mới (de novo). Tình trạng này xảy ra ngẫu nhiên trong quá trình hình thành tế bào sinh sản (trứng hoặc tinh trùng) hoặc trong quá trình phát triển sớm của thai nhi.
Các ống mật của người bệnh có thể bị hẹp, dị dạng và giảm số lượng (ống mật ít) khiến mật tích tụ trong gan và gây ra sẹo gan (xơ gan). Gan sẽ cố gắng tự phục hồi sau mỗi lần bị tổn thương. Tuy nhiên, khi tổn thương tái diễn nhiều lần khiến gan dần suy yếu, theo thời gian dẫn đến suy gan – một tình trạng rất nguy hiểm.
Triệu chứng của hội chứng Alagille
Hội chứng Alagille gây ra bất thường ở gan, tim, mắt, bộ xương, thận và các hệ cơ quan khác trong cơ thể. Triệu chứng của hội chứng Alagille thường xuất hiện trong hai năm đầu đời, rõ ràng nhất là ba tháng đầu đời, có thể nhẹ hoặc nặng tùy từng bệnh nhi. Bệnh thường ổn định khi trẻ 4-10 tuổi nhờ vào các triệu chứng được cải thiện.
Triệu chứng về da và gương mặt:
Người mắc các bệnh liên quan đến mật, gan thường có các triệu chứng về da bao gồm:
– Tắc nghẽn dòng mật từ gan (ứ mật) khiến mật thấm vào máu gây ra vàng da, mắt (lòng trắng mắt) vàng. Khoảng 90% người bệnh ống mật trong gan giảm hoặc quá ít ống mật.
– Da ngứa.
– Da thường nổi mụn mỡ.
– Tích tụ cholesterol trong da.
– Các đặc điểm trên gương mặt như mắt sâu, hai mắt cách xa nhau, trán rộng và cao, mũi thẳng, cằm nhỏ. Khoảng 90% trẻ mắc hội chứng này có dị tật mắt.

Lượng muối mật tích tụ dưới da cao có thể gây ngứa (Nguồn: Freepik)
Triệu chứng ở các cơ quan khác:
– Phân màu nhạt.
– Nước tiểu sẫm màu.
– Lá lách to.
– Gan to bất thường.
– Chậm tăng trưởng.
– Nhiễm toan ống thận.
– Bất thường về cấu trúc thận.
– Chậm hấp thu, chậm phát triển, thiếu hụt chất dinh dưỡng do cơ thể không thể hấp thu được chất chất béo và vitamin tan trong chất béo bình thường.
– Tiếng thổi ở tim (là những âm thanh xuất hiện trong chu kỳ nhịp tim do dòng máu chảy không đều trong tim).
– Khuyết tật tim bẩm sinh.
– Hẹp động mạch phổi (suy giảm lưu lượng máu từ tim vào phổi). Hẹp động mạch phổi có thể xảy ra cùng với một lỗ giữa hai buồng dưới của tim (thông liên thất) và các bất thường khác về tim. Sự kết hợp của các khuyết tật tim này được gọi là tứ chứng Fallot. Nói cụ thể hơn tứ chứng Fallot bao gồm sự kết hợp của bốn khuyết tật tim khác nhau bao gồm thông liên thất, tắc nghẽn dòng máu từ tâm thất phải đến phổi do sự thu hẹp bất thường của lỗ thông giữa động mạch phổi và tâm thất phải của tim (hẹp phổi).
– Các mạch máu trong não và tủy sống (hệ thần kinh trung ương) và thận cũng có thể bị ảnh hưởng.
– Dị tật bẩm sinh ở cột sống như đốt sống hình con bướm.
– Tầm vóc nhỏ.
Chẩn đoán hội chứng Alagille
Ngoài xem xét các triệu chứng bên ngoài, các xét nghiệm có thể được sử dụng để chẩn đoán hội chứng này bao gồm:
– Siêu âm bụng.
– Sinh thiết gan (lấy mảnh bệnh phẩm từ gan để chẩn đoán bệnh).
– Xét nghiệm chức năng thận.
– Xét nghiệm di truyền.
– Chụp X-quang có thể phát hiện bất thường ở cột sống ở một số người bệnh.

Hình ảnh mô tả gan khỏe mạnh, gan nhiễm mỡ, xơ gan (Nguồn: Freepik)
Hội chứng Alagille có các triệu chứng gần giống với các tình trạng như teo đường mật ngoài gan, ứ mật trong gan, viêm gan sơ sinh, hội chứng DiGeorge… nên có thể chẩn đoán nhầm hoặc bỏ sót.
Điều trị hội chứng Alagille
Người mắc hội chứng Alagille có thể sống bình thường nhưng trường hợp nặng như xơ gan, suy gan, tắc nghẽn mạch máu trong tim, đột quỵ (do tắc nghẽn mạch máu não)… có thể rất nguy hiểm.
Mục tiêu điều trị là giải phóng mật khỏi gan, loại bỏ tình trạng ứ mật, ngăn chặn hoặc làm chậm quá trình suy gan diễn ra. Điều trị bằng thuốc uống, thuốc thoa, phẫu thuật hoặc ghép gan có thể được sử dụng phù hợp cho bệnh nhân từ thể nhẹ đến nặng.
Thuốc uống, thuốc thoa giúp giảm cảm giác ngứa da, tăng lưu lượng mật ra khỏi gan.
Thông thường, các bác sĩ không thực hiện phẫu thuật để tái tạo ống mật chủ vì mật vẫn có thể chảy từ gan xuống ruột và hiện tại không có phương pháp nào có thể khắc phục những tổn thương trong gan.
Với những ca dị tật tim, bác sĩ có thể cân nhắc mổ để chỉnh sửa những bất thường ở cơ quan này.
Các biến chứng như suy gan, suy thận, bệnh tim rất nguy hiểm, đe doạ tính mạng của người bệnh. Khoảng 15% trường hợp bệnh gan tiến triển dẫn đến sẹo gan (xơ gan) và suy gan. Ghép gan cho bệnh nhân hội chứng Alagille sẽ được bác sĩ cân nhắc khi suy gan nặng và gan mất chức năng khiến người bệnh rơi vào tình trạng nguy hiểm. Điều đặc biệt của gan là cơ quan duy nhất trong có thể tái sinh nếu như bị mất đi một phần.
Các tình trạng bệnh thận, bất thường về đốt sống cũng được bác sĩ cân nhắc để đưa ra phương pháp điều trị phù hợp.
Nếu người bệnh bị thiếu hụt chất dinh dưỡng có thể bổ sung vitamin và các khoáng chất theo chỉ định của bác sĩ. Người bệnh cũng lưu ý ăn uống cân bằng, lành mạnh với nhiều thực phẩm bổ dưỡng để cơ thể có nhiều năng lượng, tăng sức đề kháng.
Điều trị hội chứng Alagille sớm có thể phòng ngừa và giảm biến chứng để nâng cao chất lượng cuộc sống cho người bệnh. Nếu gia đình có người mắc hội chứng này, các chuyên gia di truyền khuyến cáo nên thực hiện các xét nghiệm khi mang thai, sinh con.
Cách phát hiện sớm hội chứng Alagille
Tại thị trường Việt Nam hiện nay, Viện Di truyền Y học – Gene Solutions là đơn vị đầu tiên phát triển thành công xét nghiệm NIPT – triSure Procare sàng lọc toàn diện các bất thường di truyền phổ biến trong thai kỳ. Cụ thể gồm 27 bất thường số lượng nhiễm sắc thể cho thai, 01 mất đoạn phổ biến duy nhất được khuyến cáo sàng lọc liên quan hội chứng DiGeorge cho thai, 7.000 đột biến gây bệnh liên quan 25 bệnh đơn gen trội phổ biến nhất cho thai, 2.800 đột biến gây bệnh liên quan 9 bệnh di truyền lặn đơn gen cho mẹ. Đáng lưu ý, xét nghiệm có ý nghĩa quan trọng khi có thể phát hiện được các đột biến gen mới (de novo) gây bệnh nặng và xuất hiện ngẫu nhiên trong quá trình hình thành bào thai, không di truyền từ cha mẹ sang con hay tiền căn gia đình.
Thời điểm thai phụ có thể thực hiện xét nghiệm triSure Procare là khi thai 9 tuần tuổi, chỉ với một lần thu máu duy nhất của người mẹ.

Danh sách 25 bệnh đơn gen trội thường gặp mà gói triSure Procare sàng lọc
Các đối tượng được khuyên nên thực hiện triSure Procare gồm: thai phụ có nguy cơ cao như chồng lớn hơn 40 tuổi, gia đình có người bị rối loạn di truyền, siêu âm có bất thường như độ mờ da gáy tăng đơn độc trong quý 1 thai kỳ <3.5mm nhưng không muốn làm xét nghiệm xâm lấn, tiền căn gia đình mắc bệnh di truyền trong các gen được khảo sát.
Tư vấn di truyền
Hội chứng Alagille được di truyền theo kiểu trội trên nhiễm sắc thể thường. Khoảng 30%-50% trường hợp là do đột biến di truyền từ ba mẹ và khoảng 50%-70% trường hợp do biến thể de novo. Các trường hợp do đột biến khảm tế bào mầm hoặc tế bào sinh dưỡng của cha mẹ đã được báo cáo. Tuổi cha cao được chứng minh là có liên quan đến với các biến thể gây bệnh của hội chứng này. Nếu cha và mẹ xét nghiệm không có đột biến gen gây bệnh (thai/trẻ mắc bệnh mang đột biến de novo) thì tỉ lệ có con lần sau mang đột biến gen tăng khoảng 1-2%, do thể khảm khu trú tế bào sinh dục cần sàng lọc hoặc chẩn đoán trước sinh.
Ba hoặc mẹ bị bệnh có 50% khả năng sanh con bị bệnh. Với gia đình có tiền căn mắc hội chứng Alagille và đã xác định được đột biến gây bệnh thì có thể thực hiện xét nghiệm tiền sản cho thai phụ hoặc xét nghiệm gen cho phôi trước khi chuyển phôi. Hội chứng Alagille có mức độ biểu hiện rất khác nhau, nên mức độ biểu hiện lâm sàng không thể dự đoán được bằng xét nghiệm di truyền phân tử trước khi sinh.
Nguồn tham khảo:
1. Hopkins Medicine, Alagille Syndrome, Retrieved from
https://www.hopkinsmedicine.org/health/conditions-and-diseases/alagille-syndrome#:~:text=Alagille%20syndrome%20is%20a%20genetic,%2C%20kidney%2C%20and%20eye%20abnormalities.
2. Medlineplus, Alagille syndrome. Retrieved from
https://medlineplus.gov/genetics/condition/alagille-syndrome/
3. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Definition & Facts for Alagille Syndrome. Retrieved from
https://www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome/definition-facts
4. National Organization for Rare Disorders, Alagille Syndrome. Retrieved from
https://rarediseases.org/rare-diseases/alagille-syndrome/
5. BMC, Peritoneal dialysis in an adult patient with tetralogy of Fallot diagnosed with incomplete Alagille syndrome. Retrieved from
https://bmcmedgenet.biomedcentral.com/articles/10.1186/s12881-020-01134-7