No products in the cart.






Bệnh tan máu bẩm sinh Thalassemia thể Beta, thường gọi là bệnh Beta Thalassemia là bệnh di truyền gen lặn trên nhiễm sắc thể thường (NST số 11). Trong đó, cơ thể không sản xuất đủ lượng beta globin như bình thường. Huyết sắc tố quan trọng Hemoglobin là thành phần cơ bản của hồng cầu gồm bốn chuỗi globin (hai chuỗi alpha globin và hai chuỗi beta globin) với tỷ lệ 1/1. Chức năng chính của hồng cầu là mang oxy đến các cơ quan trong cơ thể và tham gia vào quá trình tổng hợp globin.
Bệnh Beta Thalassemia xuất phát từ đột biến trên hai gen beta globin, nằm trên nhiễm sắc thể 11. Hiện nay, đã phát hiện hơn 250 loại đột biến Beta Thalassemia, trong đó có 20 loại phổ biến. Mức độ nghiêm trọng của bệnh phụ thuộc vào loại đột biến và mức độ giảm sản xuất beta globin, có thể là giảm sự sản xuất (β+ Thalassemia) hoặc không sản xuất hoàn toàn (β0 Thalassemia).
Tan máu bẩm sinh Thalassemia thể Beta có mối liên hệ chặt chẽ với nguồn gốc dân tộc và thường xuất hiện ở các khu vực địa dư trên toàn thế giới. Theo Liên đoàn Thalassemia quốc tế (năm 2005), ước tính khoảng 1,5% dân số toàn cầu, tương đương 80-90 triệu người, mang gen Beta Thalassemia. Mỗi năm, có khoảng 60.000 trường hợp mới sinh ra mang gen bệnh này. Đặc biệt, tại khu vực Đông Nam Á, tỷ lệ người mang gen Beta Thalassemia lên đến 50% tổng số người mang gen này trên toàn thế giới, ước tính khoảng 40 triệu người. Ở Việt Nam, bệnh Beta Thalassemia phổ biến hơn ở các dân tộc ít người ở miền Bắc.
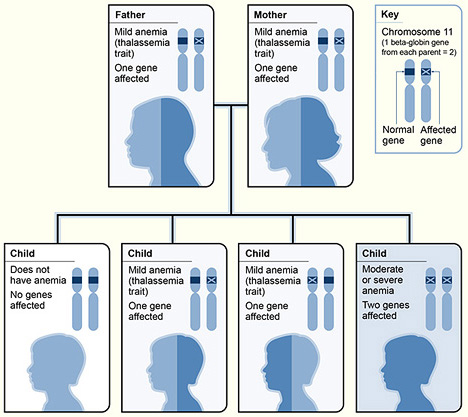
Cách di truyền bệnh Beta Thalassemia (Nguồn: wikipedia)
Phân loại các dạng bệnh tan máu bẩm sinh Thalassemia thể Beta như sau:
Thể ẩn, dị hợp tử:
Trong trường hợp này gọi là người mang gene thể ẩn trong đó một gen bị đột biến và một gen bình thường.
Thường không biểu hiện triệu chứng và chỉ thiếu máu nhẹ.
Tế bào hồng cầu thường có kích thước nhỏ. Điều này có thể làm cho tế bào hồng cầu không đáp ứng tốt với việc cung cấp sắt.
Bệnh có thể di truyền cho con cháu, do đó cần quan tâm đến khả năng truyền gen.
Thể trung gian:
Người bệnh mang hai gen bất thường, dẫn đến thiếu máu từ nhẹ đến trung bình.
Triệu chứng sớm bao gồm da nhợt nhạt, dễ cáu kỉnh, và thiếu hứng thú trong việc ăn uống.
Bệnh nhân có nguy cơ cao bị nhiễm trùng.
Quá trình điều trị phụ thuộc vào sự tiến triển của bệnh. Trong trường hợp bệnh Beta Thalassemia trung gian, nếu hàm lượng hemoglobin (Hb) trong máu thấp hơn 7 hoặc 8 mg/dl, có thể gây ra các vấn đề nghiêm trọng như kém phát triển, khó tăng cân, vàng da, vàng mắt và sự xuất hiện của màng nhầy do tăng bilirubin trong máu, cũng như nguy cơ nhiễm trùng.
Thể nặng:
Đây là dạng bệnh nghiêm trọng nhất trong bệnh Beta Thalassemia.
Người bệnh mang hai gen bất thường, dẫn đến sự suy giảm nghiêm trọng hoặc hoàn toàn thiếu hụt sản xuất beta globin, gây ra lượng hemoglobin A (Hb A) không đủ.
Bệnh thường xuất hiện ở giai đoạn đầu đời và gây ra nhiều vấn đề nghiêm trọng như thiếu máu, tăng trưởng kém, biến dạng xương nghiêm trọng.
Trẻ sơ sinh mắc bệnh thường không phát triển bình thường và không tăng cân như trẻ khác.
Biểu hiện các vấn đề về ăn uống, tiêu hóa, tăng kích thước gan, biến dạng ở xương mũi và hàm răng trên, bọng mắt và mắt xếch theo kiểu mongoloid.
Người bệnh cần thường xuyên được truyền máu và theo dõi quá trình điều trị thải sắt để giảm sắt dư thừa trong cơ thể.
Các biến chứng nguy hiểm mà Thalassemia thể Beta có thể gây ra đòi hỏi sự quan tâm đặc biệt:
Phình to lách, gan và tim:
Nếu không tiến hành điều trị hiệu quả, lách, gan và tim của người bệnh có xu hướng tăng kích thước.
Trong trường hợp lách quá to, có thể cần phải thực hiện phẫu thuật cắt bỏ lách để giảm đau và khó chịu cho bệnh nhân.
Tuy nhiên, việc loại bỏ lách có thể tạo điều kiện thuận lợi cho sự phát triển của nhiễm khuẩn, đặc biệt là sau khi thực hiện phẫu thuật.
Vấn đề về xương:
Xương thường trở nên mỏng, giòn, và biến dạng.
Bệnh nhân cần tiến hành truyền máu thường xuyên để duy trì sự sống. Tuy nhiên, việc truyền máu có thể tăng nguy cơ tích tụ sắt trong tim và các cơ quan khác. Sắt tích tụ có thể gây ra suy tim sớm, thậm chí ở giai đoạn thiếu niên, là một biến chứng nguy hiểm khác của tan máu bẩm sinh Thalassemia thể Beta.
Theo dõi và điều trị thích hợp bởi các bác sĩ chuyên khoa là rất quan trọng để ngăn ngừa và quản lý các biến chứng của bệnh tan máu bẩm sinh Thalassemia thể Beta.
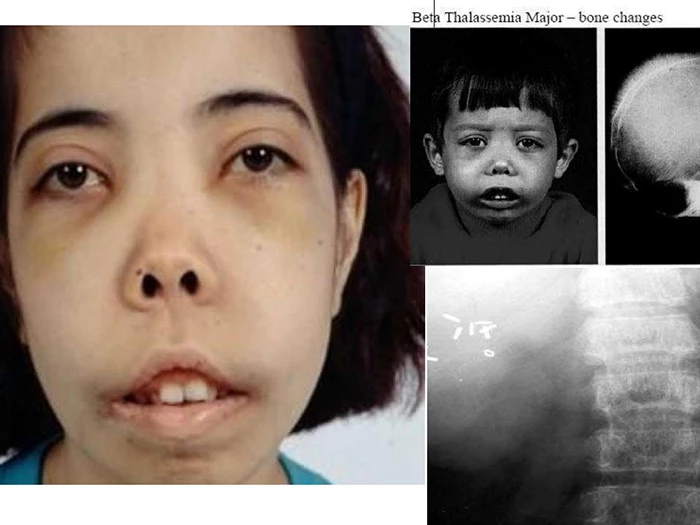
Gương mặt điển hình của người bệnh Thalassemia thể Beta (Nguồn: wecapable)
Bệnh tan máu bẩm sinh Thalassemia thể Beta xuất phát từ các đột biến trên gen HBB, đây là nguyên nhân chính dẫn đến bệnh. Gen Thalassemia mang thông tin để tổng hợp protein được gọi là beta-globin, một thành phần quan trọng của hemoglobin – huyết tương có nhiệm vụ chứa và vận chuyển oxy trong hồng cầu. Hemoglobin bao gồm bốn đơn vị, trong đó có hai đơn vị beta-globin và hai đơn vị alpha-globin khác.
Có một số loại đột biến trên gen HBB có tác động tiêu cực lên quá trình sản xuất beta-globin. Trong một số trường hợp, đột biến này có thể ngăn chặn hoàn toàn việc sản xuất beta-globin, dẫn đến tình trạng Thalassemia beta-zero. Trong trường hợp khác, đột biến cho phép sản xuất beta-globin, nhưng với số lượng giới hạn, tạo ra tình trạng Thalassemia beta-plus.
Thiếu beta-globin dẫn đến giảm lượng hemoglobin chức năng trong hồng cầu, dẫn đến thiếu hụt hồng cầu trưởng thành. Sự thiếu hụt này gây ra hiện tượng thiếu máu và nhiều vấn đề sức khỏe liên quan đến Thalassemia thể Beta.
Sàng lọc trước sinh
Các cặp đôi chuẩn bị sinh con hoặc thai phụ có thể chọn làm xét nghiệm CarrierThalass. CarrierThalass được sử dụng công nghệ giải trình tự gen thế hệ mới kết hợp Gap-PCR giúp tối ưu phạm vi khảo sát, phát hiện 495 mất đoạn và đột biến gây bệnh Thalassemia Alpha, Beta, khảo sát được các mất đoạn gây bệnh SEA, 3.7, 4.2, THAI phổ biến nhất. Xét nghiệm có thể đánh giá trực tiếp các bất thường di truyền là nguyên nhân gây bệnh, xác định chính xác kiểu gen của cha mẹ và tính toán nguy cơ mang gen bệnh cho con trong tương lai.
Đặc biệt, Gene Solutions tiên phong phát triển xét nghiệm NIPT triSure Thalass có phạm vi khảo sát tích hợp vừa sàng lọc các dị tật bẩm sinh phổ biến như Down, Edwards, Patau, Turner và vừa phát hiện bệnh Thalassemia cho thai phụ theo chương trình mục tiêu dân số quốc gia (Nghị quyết số 21-NQ/TW). Nhằm đạt được mục tiêu hiệu quả và kinh tế, phương pháp phân tích đột biến Thalassemia dựa trên DNA ngoại bào của thai phụ và thuật toán phân tích đã được kiểm chứng độ chính xác cao > 99%.

Xét nghiệm NIPT tích hợp sàng lọc bệnh Thalassemia là một giải pháp thiết thực, mang đến những lợi ích vượt trội so với chi phí bỏ ra ban đầu, với tỷ lệ ước tính 73:1. Chỉ 1 lần lấy máu duy nhất, thai phụ có thể thực hiện triSureThalass để sàng lọc dị tật bẩm sinh phổ biến của thai nhi, cũng như xét nghiệm tầm soát bệnh Thalassemia thể alpha và beta cho người mẹ. Trường hợp thai phụ làm xét nghiệm phát hiện mang gen bệnh thì người chồng cũng cần làm xét nghiệm kiểm tra. Nếu cả bố và mẹ cùng mang gen của 1 thể thalassemia, thì tùy vào việc bố mẹ mang gen thể gì để bác sĩ tư vấn cho thai phụ làm chẩn đoán trước sinh hoặc sau sinh. Đối với các thể nặng và rất nặng, thai phụ sẽ được tiến hành sinh thiết gai nhau (thai 11-13 tuần) hoặc chọc ối (thai > 15 tuần) và hỗ trợ xét nghiệm gen chẩn đoán CarrierThalass miễn phí.
Sàng lọc sau sinh
Phương pháp sàng lọc và chẩn đoán bệnh Thalassemia thể Beta đang được thực hiện một cách có tổ chức và hiệu quả tại Việt Nam. Theo Bệnh viện và Trung tâm Truyền máu Huyết học Trung ương, ước tính có khoảng 13 triệu người Việt Nam mang gen đột biến Thalassemia thể ẩn. Do đó, tất cả các trẻ sơ sinh tại bệnh viện được tiến hành sàng lọc bằng cách lấy mẫu máu từ gót chân. Khi kết quả sàng lọc cho thấy nguy cơ cao về bệnh liên quan đến hemoglobin, trẻ sẽ được lấy mẫu để thực hiện xét nghiệm gen chẩn đoán bệnh Thalassemia. Điều này giúp bác sĩ có thông tin cụ thể về gen của trẻ, từ đó xác định phương án điều trị thích hợp và kịp thời, giúp giảm thiểu các tác động tổn hại trong trường hợp trẻ mắc phải bệnh Thalassemia nặng.

Trẻ nhỏ sẽ bị thiếu máu nặng gây nguy hiểm đến tính mạng (Ảnh: Freepik)
Trong việc điều trị bệnh tan máu bẩm sinh Thalassemia thể Beta, hiện nay có hai phương pháp chính được áp dụng, bao gồm truyền máu và thải sắt. Tùy thuộc vào tình trạng của từng bệnh nhân, cũng có một số biện pháp điều trị khác được áp dụng để đảm bảo hiệu quả điều trị tốt nhất.
Truyền máu:
Bệnh nhân Thalassemia thể Beta thường mắc thiếu máu mạn tính và cần truyền máu định kỳ.
Mục tiêu của truyền máu là duy trì nồng độ huyết sắc tố khoảng 90-100g/l (tùy thuộc vào mức độ nghiêm trọng của bệnh).
Khoảng cách giữa các lần truyền máu thường là từ 2-5 tuần và sử dụng chế phẩm máu là hồng cầu khối, không sử dụng máu toàn phần.
Thải sắt:
Thải sắt được thực hiện để ngăn ngừa quá tải sắt, hạn chế biến chứng trên các tổ chức và cơ quan trong cơ thể. Bệnh nhân thường phải duy trì việc sử dụng thuốc thải sắt suốt đời.
Cắt lách:
Thủ thuật cắt lách được xem xét trong các trường hợp như: tăng nhu cầu truyền máu, lách quá to gây đau, cản trở hoạt động hàng ngày, hoặc gây ra cường lách, dẫn đến giảm bạch cầu hoặc tiểu cầu.
Ghép tế bào gốc:
Đây là một phương pháp điều trị hiện đại và có tiềm năng chữa khỏi bệnh, nhưng khả năng tìm người phù hợp cho cấy ghép là thấp và chi phí điều trị rất cao.
Với những bệnh nhân có biến chứng nặng ở các bộ phận như gan, tim, tỷ lệ thành công sẽ thấp hơn.
Chăm sóc toàn diện:
Chăm sóc toàn diện đóng vai trò quan trọng trong việc phòng ngừa và hạn chế các biến chứng của bệnh, giúp cải thiện chất lượng cuộc sống của bệnh nhân.
Điều trị biến chứng:
Đối với những biến chứng như suy tuyến nội tiết, đái tháo đường, suy tim, xơ gan, loãng xương, rối loạn đông máu, điều trị được đặt ra dựa trên tình trạng cụ thể của bệnh nhân.
Tính di truyền của bệnh tan máu bẩm sinh Thalassemia thể Beta được giải thích như sau:
Beta thalassemia là một bệnh di truyền theo kiểu lặn (recessive), điều này có nghĩa là để phát triển bệnh, cần phải có đột biến trên cả hai bản sao của gen HBB trong mỗi tế bào.
Cha mẹ của người bệnh thường mang một bản sao của gen đột biến HBB, nhưng không biểu hiện bất kỳ triệu chứng hay dấu hiệu của bệnh. Trong một số trường hợp, người mang chỉ một bản sao của gen đột biến HBB có thể gặp thiếu máu nhẹ và được xem là thể nhẹ của Thalassemia.
Tan máu bẩm sinh Thalassemia thể Beta là bệnh di truyền lặn, tức cha mẹ bình thường vẫn có thể mang gen đột biến và di truyền cho con. Tỷ lệ con sinh ra mang gen bệnh lặn và có biểu hiện bệnh là 25%. Nguy cơ là như nhau đối với nam và nữ.
Phòng ngừa bệnh Thalassemia thể Beta là một phần quan trọng trong quản lý sức khỏe di truyền. Bệnh này thường không có biểu hiện rõ ràng ở người mang gen đột biến, và một số người chỉ biết sau khi có con mắc bệnh. Vì vậy, việc xét nghiệm gen lặn trước khi mang thai là cách quan trọng để ngăn ngừa bệnh tan máu bẩm sinh Thalassemia thể Beta. Điều này giúp đảm bảo rằng con cái được sinh ra là khỏe mạnh và không bị bệnh Thalassemia.
Chăm sóc người mắc bệnh tan máu bẩm sinh Thalassemia thể Beta đòi hỏi sự quan tâm và thực hiện các biện pháp cụ thể như sau:
Đối với những người mắc Thalassemia ở thể trung gian hoặc nặng:
Quan trọng là phải thực hiện kiểm tra y tế định kỳ và nếu có ý định kết hôn, cần thực hiện kiểm tra tiền hôn nhân để xác định xem bạn đời có mang gen bệnh tương tự hay không.
Trong trường hợp cả hai vợ chồng đều mang gen bệnh, cần lập kế hoạch hợp lý để có con khỏe mạnh, có thể cần tư vấn với chuyên gia di truyền.
Đối với phụ nữ mang bệnh và điều trị truyền máu nhiều lần:
Cần kiểm tra mức sắt dư thừa trong cơ thể trước khi quyết định có thai, vì lượng sắt dư thừa cao có thể ảnh hưởng đến sự phát triển của thai nhi trong giai đoạn đầu của thai kỳ.
Khi khó mang thai tự nhiên:
Đôi khi, bệnh Thalassemia có thể làm cho việc mang thai tự nhiên trở nên khó khăn hơn. Trong trường hợp này, hãy tìm đến các bác sĩ chuyên khoa hỗ trợ sinh sản để kiểm tra, tư vấn và hỗ trợ.
Phụ nữ nên bắt đầu uống axit folic ít nhất 3 tháng trước khi có ý định mang thai và cần được kiểm tra thai kỳ thường xuyên để theo dõi các vấn đề như thiếu máu, suy tim, suy gan, quá tải sắt…
Chăm sóc tổng quan:
Người bệnh Thalassemia, đặc biệt là những người mang thể nặng, cần duy trì lối sống lành mạnh và tuân thủ lịch hẹn kiểm tra y tế định kỳ theo chỉ định của bác sĩ.
Cần hạn chế sử dụng các chất bổ sung chứa sắt hoặc vitamin khi chưa được chỉ định bởi bác sĩ.
Chế độ ăn uống cần cân đối và hạn chế thực phẩm giàu chất sắt nếu lượng sắt trong máu đã cao.
Nếu cần, bác sĩ có thể đề nghị bổ sung axit folic vào chế độ dinh dưỡng hàng ngày.
Rửa tay và duy trì vệ sinh cá nhân thường xuyên để tránh nhiễm trùng, đặc biệt quan trọng đối với những người đã thực hiện cắt lá lách.
Cần tuân thủ tiêm phòng và tư vấn y tế định kỳ để duy trì sức kháng.
Beta Thalssemia di truyền theo kiểu gen lặn trên nhiễm sắc thể thường.
Trẻ mắc bệnh di truyền lặn (trên NST thường) chỉ biểu hiện triệu chứng khi mang cả 2 bản sao của gen đột biến được thừa hưởng từ bố và mẹ. Trường hợp bố và mẹ hoàn toàn khỏe mạnh nhưng là người lành mang gen bệnh vẫn có thể sinh con mắc di truyền lặn.
Khả năng di truyền cho con như sau:
– 25% khả năng sinh con mắc bệnh Beta Thalassemia
– 50% khả năng sinh con là người lành mang gen bệnh
– 25% khả năng sinh con bình thường không mang gen bệnh
Nguồn tham khảo:
https://medlineplus.gov/genetics/condition/beta-thalassemia/
https://www.hopkinsmedicine.org/health/conditions-and-diseases/beta-thalassemia
https://kidshealth.org/en/parents/beta-thalassemia.html
https://familydoctor.org/condition/thalassemia/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8683628/

Chương trình dành cho cộng đồng “Vì một Việt Nam không còn nỗi lo ung thư”, nhằm góp phần nâng cao nhận thức về tầm soát, phát hiện sớm ung thư thông qua hình thức tặng 7.000 suất tầm soát ung thư miễn phí bằng công nghệ SPOT-MASTM, cho hơn 100 doanh nghiệp, bệnh viện và phòng khám khắp cả nước.
» Xem chi tiết «
» Xem trên VNExpress «



