









Phenylketon niệu là một rối loạn chuyển hóa di truyền hiếm gặp, gây ra dư thừa một axit amin gọi là phenylalanine. PKU được gây ra bởi sự thay đổi gen phenylalanine hydroxylase (PAH). Gen này giúp tạo ra enzyme cần thiết để chuyển hóa phenylalanine. Nếu không có enzyme cần thiết để chuyển hóa phenylalanine, dẫn đến sự tích lũy phenylalanine trong cơ thể, khi một người bị PKU ăn thực phẩm có chứa protein hoặc ăn aspartame, một chất làm ngọt nhân tạo. Điều này cuối cùng có thể dẫn đến các vấn đề sức khỏe nghiêm trọng.

Tổng quan về bệnh Phenylketon niệu (Nguồn: Pinterest)
Trẻ sơ sinh bị Phenylketon niệu ban đầu không có bất kỳ triệu chứng nào. Tuy nhiên, nếu không điều trị, trẻ sơ sinh thường phát triển các dấu hiệu của Phenylketon niệu trong vòng vài tháng. Các dấu hiệu và triệu chứng của Phenylketon niệu không được điều trị có thể nhẹ hoặc nặng, bao gồm:
– Mùi mốc trong hơi thở, da hoặc nước tiểu, gây ra bởi quá nhiều phenylalanine trong cơ thể
– Các vấn đề về hệ thần kinh như co giật, chậm phát triển tâm thần vận động
– Phát ban da, chẳng hạn như bệnh chàm
– Màu da, tóc và mắt sáng hơn so với các thành viên trong gia đình, bởi vì phenylalanine không thể biến đổi thành melanin – sắc tố chịu trách nhiệm cho tóc và màu da
– Kích thước đầu nhỏ bất thường (microcephaly)
– Tăng động
– Khuyết tật trí tuệ
– Chậm phát triển
– Các vấn đề về hành vi, cảm xúc và xã hội
– Rối loạn sức khỏe tâm thần
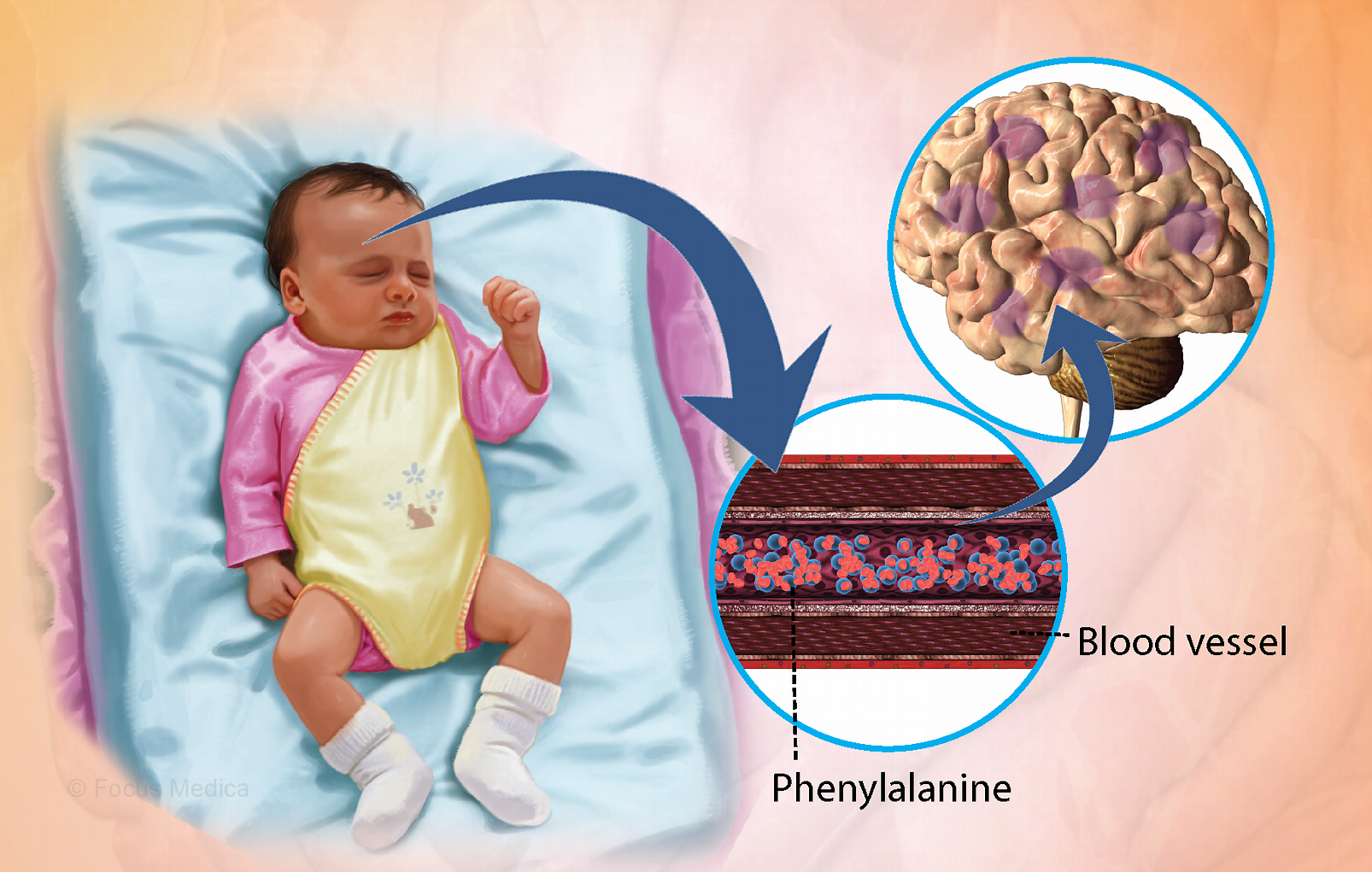
Trẻ bị bệnh Phenylketon niệu thường gặp vấn đề về trí tuệ (Nguồn: MSN)
Kiểu hình bệnh đa dạng, từ thể cổ điển, thể trung bình, thể nhẹ tới thể chỉ có tăng Phenylalanin lành tính không triệu chứng, tùy thuộc vào hoạt độ enzyme còn lại.
Phenylketon niệu cổ điển: Đây là dạng nghiêm trọng nhất của bệnh. Enzym cần thiết để chuyển hóa phenylalanine bị thiếu hoặc giảm nghiêm trọng. Điều này dẫn đến nồng độ phenylalanine cao có thể gây tổn thương não nghiêm trọng.
Các dạng Phenylketon niệu ít nghiêm trọng hơn: Ở dạng nhẹ hoặc trung bình, enzyme vẫn còn một phần chức năng, vì vậy nồng độ phenylalanine không cao, dẫn đến nguy cơ tổn thương não thấp hơn. Bất kể thể bệnh nào, hầu hết trẻ sơ sinh, trẻ em và người lớn mắc bệnh vẫn cần chế độ ăn uống tiết chế protein đặc biệt để ngăn ngừa biến chứng khuyết tật trí tuệ và các biến chứng khác.
Phụ nữ mang thai bị Phenylketon niệu
Phụ nữ bị Phenylketon niệu và mang thai có nguy cơ mắc một dạng khác của bệnh gọi là PKU của mẹ. Nếu phụ nữ mắc bệnh không tuân theo chế độ ăn kiêng PKU đặc biệt trước và trong khi mang thai, nồng độ phenylalanine trong máu có thể trở nên cao và gây hại cho em bé đang phát triển. Ngay cả những phụ nữ mắc bệnh PKU thể nhẹ cũng có thể khiến thai nhi của họ gặp nguy hiểm nếu không tuân theo chế độ ăn kiêng PKU. Khi sinh ra, em bé có thể bị nhẹ cân, tật đầu nhỏ, tim bẩm sinh. Ngoài ra, PKU của mẹ có thể khiến trẻ chậm phát triển, thiểu năng trí tuệ và các vấn đề về rối loạn hành vi.
Để cải thiện chất lượng sống cho trẻ thì bệnh nên được chẩn đoán sớm bằng xét nghiệm di truyền sàng lọc trước sinh, từ đó xây dựng kế hoạch can thiệp, điều trị kịp thời. Trong vòng 48 đến 72 giờ sau khi sinh, trẻ sẽ được lấy máu gót chân bằng giấy thấm chuyên dụng để thực hiện sàng lọc sơ sinh.
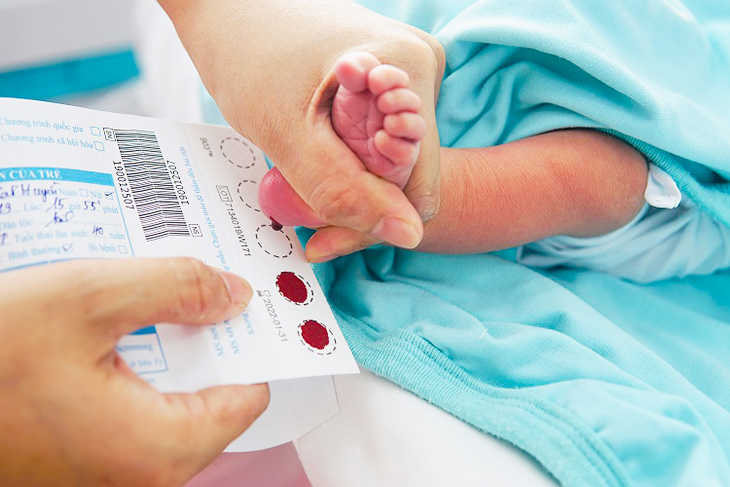
Xét nghiệm babySure là mô hình xét nghiệm đầu tiên kết hợp từ sàng lọc sinh hóa đến chẩn đoán sinh hóa và di truyền cho trẻ sơ sinh tại Việt Nam. Xét nghiệm áp dụng kỹ thuật miễn dịch enzyme huỳnh quang và công nghệ giải trình tự gen để chẩn đoán di truyền nhằm phát hiện sớm 05 bệnh lý rối loạn chuyển hóa bẩm sinh của trẻ như thiếu men G6PD, rối loạn chuyển hóa galactose, Phenylketon niệu, suy giáp bẩm sinh, tăng sản thượng thận bẩm sinh, từ đó có hướng điều trị hiệu quả và can thiệp kịp thời.
Nếu kết quả sàng lọc cho thấy nguy cơ cao với bệnh Phenylketon niệu, trẻ cần được làm xét nghiệm chẩn đoán và các xét nghiệm này sẽ được Gene Solutions hỗ trợ miễn phí.
Ngay từ những ngày đầu sau sinh bé phải được thăm khám và tư vấn điều trị bởi các chuyên gia để có chế độ dinh dưỡng hợp lý nhằm hạn chế tối đa sự biểu hiện của bệnh.
Nhìn chung điều trị Phenylketon niệu là chế độ ăn hạn chế phenylalanine suốt đời, đồng thời bổ sung vitamin và khoáng chất. Phenylalanine được tìm thấy trong tất cả các loại đạm động vật và đạm thực vật cũng như chất làm ngọt nhân tạo hay được cho vào các loại thực phẩm ăn kiêng. Trẻ phải hạn chế ăn các loại thực phẩm có hàm lượng đạm cao như: thịt, trứng, sữa, phô-mai, thay vào đó là sử dụng các loại thực phẩm có hàm lượng đạm thấp như rau, trái cây, các loại sữa đã được xử lý để tách phenylalanine.
Các bậc phụ huynh khi chuẩn bị thực phẩm cho con mình nên tập thói quen nhìn vào thành phần các chất trên nhãn đồ ăn để xem loại thực phẩm đó có phenylalanine hay không.
Điều trị PKU ở phụ nữ mang thai (được gọi là PKU của mẹ) có thể được ngăn ngừa các biến chứng cho thai bằng cách kiểm soát chế độ ăn uống của người mẹ trước và trong thai kỳ.
Sapropterin dihydrochloride tương tự BH4 tổng hợp (Kuvan®, BioMarin Corporation, Tiburon, CA), đã được FDA phê duyệt để điều trị tăng phenylalanin máu ở bệnh nhân thiếu PKU hoặc BH4, do đó một số bệnh nhân bị PKU hiện có thể bỏ qua việc điều trị bằng chế độ ăn kiêng.
Liệu pháp trị liệu mới Pegvaliase (PALYNZIQ®, BioMarin Pharmaceutical Inc., Hoa Kỳ) là một liệu pháp thay thế enzyme mới cho PKU mà FDA đã phê duyệt vào năm 2018 cho người lớn ở Hoa Kỳ và cho bệnh nhân ≥ 16 tuổi ở Châu u có nồng độ Phe trong máu không kiểm soát được > 600 mol/L. Pegvaliase là có khả năng hạ thấp mức Phe trong máu xuống mức bình thường bất kể dữ liệu về BH4 hoặc kiểu gen. Mặc dù hiệu quả nhưng các tác dụng phụ bất lợi bao gồm phản ứng trên da, đau khớp và phản ứng phản vệ hiếm gặp.

Bản chất của bệnh Phenylketon niệu là bệnh di truyền lặn trên nhiễm sắc thể thường nên có những trường hợp không biểu hiện thành bệnh mà vẫn mang gen đột biến gọi là người lành mang gen và có khả năng di truyền gen bệnh cho con. Do dó phương pháp phòng tránh hiệu quả nhất là cha mẹ nên chủ động tầm soát nguy cơ mang gen bệnh với xét nghiệm gen triSure Carrier, từ đó xác định nguy cơ mắc bệnh ở con.
Ngoài tầm soát nguy cơ mang gen bệnh Phenylketon niệu, triSure Carrier còn sàng lọc nguy cơ mang gen thêm 8 bệnh di truyền lặn phổ biến và nguy hiểm khác như: Tan máu bẩm sinh Thalassemia thể Alpha, Tan máu bẩm sinh Thalassemia thể Beta, Thiếu men G6PD, Vàng da ứ mật do thiếu men citrin, Rối loạn phát triển giới tính ở nam do thiếu men 5-alpha reductase, Bệnh Pompe (rối loạn dự trữ glycogen nhóm 2), Bệnh Wilson (rối loạn chuyển hóa đồng), Rối loạn chuyển hóa đường galactose.

Thời điểm lý tưởng nhất nên làm xét nghiệm triSure Carrier là khi dự định có con hoặc trong khi mang thai, để từ đó xác định sớm nguy cơ mắc bệnh nhằm xây dựng kế hoạch can thiệp sớm ngay sau sinh.
Hiện Gene Solutions đang áp dụng mô hình kết hợp NIPT sàng lọc dị tật di truyền và sàng lọc bệnh di truyền lặn đơn gen. Cụ thể, khi thai phụ chọn xét nghiệm triSure9.5 trở lên sẽ được thực hiện miễn phí gói triSure Carrier. Tất cả quá trình xét nghiệm này đều chỉ cần một lần lấy máu ở người mẹ.
Qua 2 bài nghiên cứu khoa học về “Combined Gap-Polymerase Chain Reaction and Targeted Next-Generation Sequencing Improve α- and β-Thalassemia Carrier Screening in Pregnant Women in Vietnam” và “Detection of maternal carriers of common α-thalassemia deletions from cell-free DNA” được đăng tải trên các tạp chí y khoa quốc tế uy tín năm 2022, Gene Solutions đã chứng minh được hiệu quả của việc sử dụng cfDNA từ NIPT để phát hiện người mẹ mang gen bệnh lặn.
Gen PAH mã hóa cho enzyme phenylalanine hydroxylase tham gia vào quá trình chuyền hóa Phenylalanin (Phe) thành Tyrosine (Tyr). Sự rối loạn này gây thiếu hụt Tyrosine – tiền chất quan trọng để sản xuất serotonin, catecholamine dẫn truyền thần kinh, melanin và hormon tuyến giáp.
Phenyketon niệu (PKU) là bệnh lý di truyền lặn trên NST thường, do đồng hợp tử/dị hợp tử phức đột biến gây bệnh trên gen PAH gây ra. PKU không đồng nhất về mặt di truyền, với hơn 1.000 biến thể PAH được báo cáo ở những cá nhân mắc PKU trên toàn thế giới. Người mắc bệnh phải nhận đồng thời 2 bản sao của gen đột biến của bố và mẹ. Trường hợp cả bố và mẹ đều là người khỏe mạnh nhưng là người lành mang gen bệnh thì nguy cơ di truyền cho con của họ như sau:
– 25% con là người hoàn toàn khỏe mạnh và không mang gen bệnh
– 50% con là người lành mang gen bệnh
– 25% con là người mắc bệnh
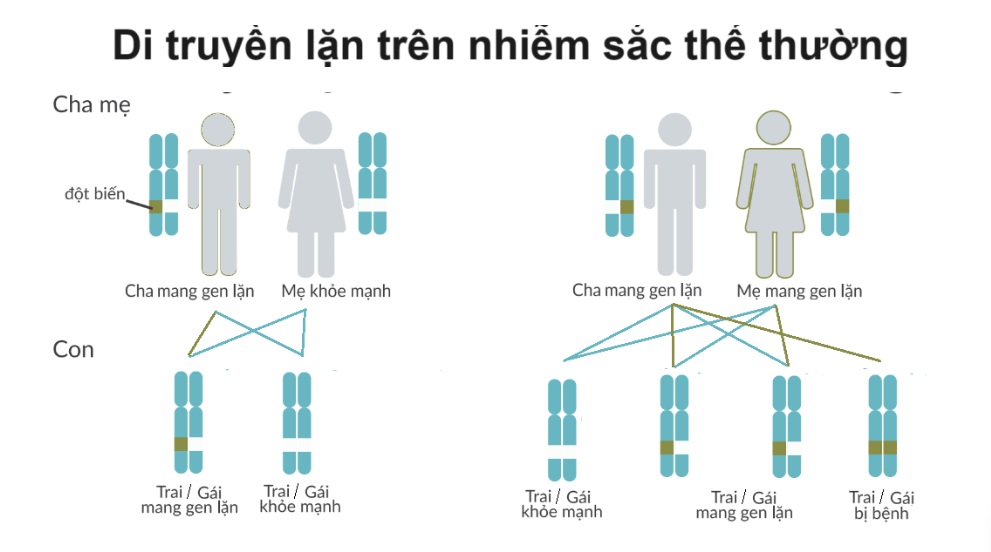
Nguyên lý di truyền gen lặn
Trường hợp bố mẹ là người lành mang gen (carrier) có khả năng sinh con mắc bệnh khi trẻ nhận 2 đột biến từ cả bố và mẹ. Trường hợp bố mẹ mang gen cần tư vấn nguy cơ sinh con mắc bệnh, tư vấn điều trị với các trường hợp chẩn đoán là người bệnh Phenylketon niệu, thai phụ mắc bệnh cần tư vấn điều trị, hạn chế các biến chứng cho thai nhi.
Nguồn tham khảo:
https://www.mayoclinic.org/diseases-conditions/phenylketonuria/symptoms-causes/syc-20376302#:~:text=Phenylketonuria%20
https://medlineplus.gov/genetics/condition/phenylketonuria/

Chương trình dành cho cộng đồng “Vì một Việt Nam không còn nỗi lo ung thư”, nhằm góp phần nâng cao nhận thức về tầm soát, phát hiện sớm ung thư thông qua hình thức tặng 7.000 suất tầm soát ung thư miễn phí bằng công nghệ SPOT-MASTM, cho hơn 100 doanh nghiệp, bệnh viện và phòng khám khắp cả nước.
» Xem chi tiết «
» Xem trên VNExpress «